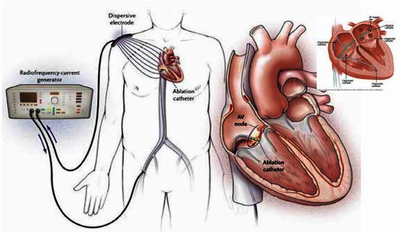
The NMPA released the “Draft Guideline on Clinical Evaluation for Cardiac Radiofrequency Ablation Devices” on July 2, 2026 for feedback. Feedbacks need to be submitted by July 30, 2026. The document provides a structured framework for manufacturers to generate clinical evidence for these high-risk Class III devices, while also serving as a reference for technical reviewers. Its significance lies in clarifying when a full clinical trial is required versus when a same-product clinical evaluation pathway may be acceptable, thus streamlining the registration process for device updates.
Currently, Boston Scientific, Johnson & Johnson, and Abbott have already registered cardiac radiofrequency ablation systems and catheters with the NMPA.
The draft guideline explicitly states that if a prior-generation product has been approved in China, and the applicant seeks to register a design-change version of that product, the applicant may use same-variety comparison pathway. Non-clinical study data, clinical data from the prior-generation product, and overseas clinical data for the new device can be used to demonstrate safety, efficacy, and performance. In this context, clinical data may derive, both domestically and internationally, from clinical trial data, clinical experience data, and clinical literature data.
For Clinical Guideline on RFA & MWA Equipment, to treat liver cancer and thyroid nodules, please click HERE
Scope of Application
This draft guideline applies to cardiac RF ablation devices and catheters used in electrophysiology to deliver RF energy to myocardial or pulmonary vein tissues, creating coagulative necrosis to treat tachyarrhythmias. Additional functionalities may include temperature sensing, impedance monitoring, contact force, magnetic navigation, and saline irrigation.
Devices fall under category 01-03-02 and catheters under 01-03-04, both Class III (high-risk).
Primary indications cover drug-refractory, recurrent, symptomatic paroxysmal AF and persistent AF (duration <1 year). Pulsed-field, cryoablation, and laser systems may reference relevant sections when seeking these same indications.
General Requirements for Clinical Trials
Trial design:
A prospective, single-arm target-value design is generally acceptable. For major design changes or entirely novel indications, a randomized controlled trial (RCT) using an approved product or standard medical therapy as control is recommended.
Patient selection & blanking period:
Inclusion requires documented drug-refractoriness (intolerance or contraindication to ≥1 Class I/III antiarrhythmic) and recurrent symptomatic episodes. A 90-day blanking period applies—events here do not count as primary failures; amiodarone is generally avoided during this window.
Primary endpoints:
Efficacy: 12-month treatment success rate (immediate procedural success + freedom from recurrence ≥30s, re-ablation, cardioversion, or use of previously ineffective Class I/III drugs or amiodarone after blanking).
Safety:
Composite major adverse events (death, MI, stroke, TIA, thromboembolism, phrenic nerve palsy, vascular/conductive/gastric issues within 7 days; tamponade/pericarditis within 30 days; pulmonary vein stenosis and atrio-esophageal fistula within 12 months).
Secondary endpoints:
Include immediate/long-term/single-procedure success, drug-free success, re-ablation rate, procedure times, contact force/impedance changes, fluoroscopy exposure, isolation proportion, ablation parameters, and quality of life.
Sample size & follow-up:
Minimum 120 subjects for paroxysmal/persistent AF applications (based on 50% and 40% success targets at 12 months). Follow-up at 7 days, 1, 3, 6, and 12 months, with a ≥90% follow-up rate. Statistical methods include descriptive analysis, hypothesis testing (one-sided α=0.025), interval estimation, and bias control, with careful handling of non-contemporaneous bias in single-arm designs.
Same-Variety Comparison Pathway
Eligibility:
Applicable if the prior-generation device from the same applicant is already approved in China, and the current application is for a design change, allowing use of existing nonclinical, prior-generation clinical, and overseas data to demonstrate compliance.
Exclusion (when a trial is still required):
If the new device introduces entirely novel materials, structural designs, or technological features that cannot be adequately bridged by existing data, a full clinical trial remains mandatory.
Comparative requirements:
The prior-generation product serves as the comparator, requiring comprehensive side-by-side comparisons of indications, patient populations, design, materials, and technical performance under identical test conditions.
Addressing differences:
Specific differences—such as contact force sensing, localization type (magnetic vs. magnetoelectric), temperature sensor configuration, irrigation port count, microelectrode additions, or deflection direction—must be rigorously supported by bench tests, animal studies, and/or other nonclinical evidence to prove no adverse impact on safety or effectiveness.
Data sources & labeling:
Clinical data for the comparator may include published literature, trial data, and clinical experience from domestic and international sources via systematic searches. The final device labeling must summarize the clinical evaluation results (study design, population, sample size, endpoints, outcomes, adverse events, and recommended settings), with the main unit labeling referencing the approved catheter instructions for compatible parameters.