
NMPA (CFDA) clinical trial requirements have been significantly strengthened over the years.
- In 2016, 8 out of 20 clinical trials have been identified with issues in clinical trial audits.
- In 2017, 3 out of 19 trials have been identified with issues.
- In 2018, 9 of 10 clinical trials have been identified (the results of another 10 trials have not been announced.)
Trends in NMPA Clinical Trial Audits
- NMPA not only audits clinical trials for registration, but also inspect those for conditional approval (already approved but more clinical research is needed).
- More IVD clinical trials to receive clinical audit. NMPA published “Draft IVD Clinical Trial Guideline for Feedback” on November 22, 2018. It specifies that IVD clinical trial sites must be listed on the NMPA (CFDA) filed institutions for Medical Device Clinical Trial Sites. In addition, it requires using fresh samples, instead of stored sample, and statistically significant sampling size.
Note: For our post on Draft Guideline on IVD Clinical Trial, please click HERE.
For Draft Guideline on IVD Clinical Trial, please email info@ChinaMedDevice.com.
- More compliance issues, less data authenticity issues. In 2018, all defected clinical trials were found with compliance issues, which means the process of clinical trial is not in compliance with NMPA regulations.
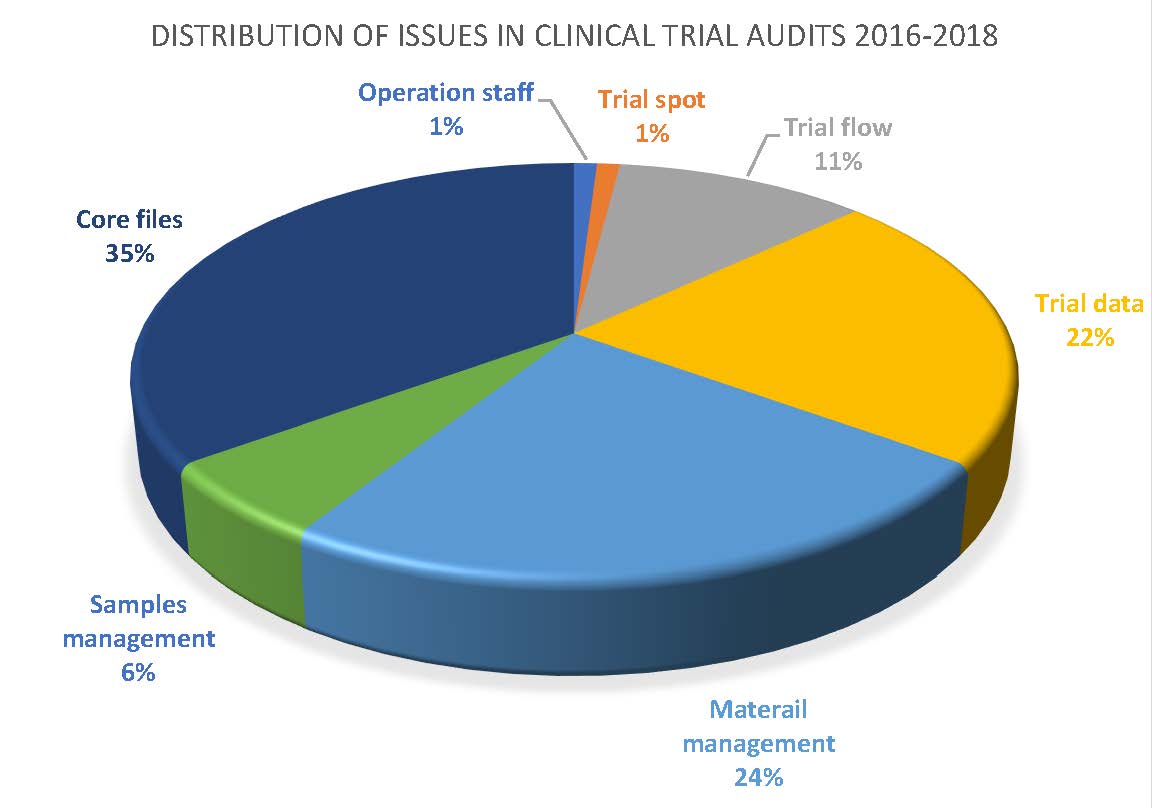
Among them, ‘core files’ issues stand for 35% of all issues found in clinical trials audits. ‘Material management’ and ‘trials data’ represent 24% and 22% respectively.
Summary of NMPA Reported Problems
Clinical trial site
- Clinical trial site is not listed on the NMPA (CFDA) filed institutions.
Note: For Catalog of 676 Registered Clinical Trial Sites NMPA issued on January 18, 2019, please contact info@ChinaMedDevice.com.
Clinical trial personnel
- Clinical trial personnel are not from the clinical trial institution.
Clinical trial protocol
- The investigator was not involve in the whole process of designing the clinical trials protocol;
- The clinical trials protocol was not in accordance with the intended use of medical device;
- The protocol was not approved by the hospital ethics committee, the sponsor or the primary investigator (PI);
- The protocol has no signature from PI, statistician, clinical site(s) or the NMPA clinical trial department.
Ethics committee
- Informed Consent Form (ICF) and Case Report Form (CRF) from Ethics committee’s Letter of Agreement are inconsistent with executed ICF and CRF;
- Ethics committee members’ opinion was not provided;
- Letter of Agreement did not contain members’ attendee form;
- Letter of Agreement did not contain version number of clinical trial protocol and ICF.
- ICF did not contain Patient Warnings;
- ICF was signed by patients’ relative without plausible reason.
Clinical trial Preparation
- There were no technical indicators of tested products in the ‘Medical Device Clinical Test Instructions’;
- The cases were not selected according to statistical principles, and the samples were not traceable;
- The standard operating procedures (SOPs) were not formulated according to the clinical trial protocol;
- Clinical trial quality control transfer & inspection records were not provided;
- The relevant qualification documents of the control products were not provided;
- In the multi-center test, the reference reagents were different;
- The clinical trial records, forms, documents and other materials had not been signed or sealed.
Clinical trial implementation
- The clinical trial procedures were inconsistent with the clinical trial protocol;
- The test of subjects was incomplete before enrolling;
- Preliminary experiments were not conducted;
- The audit protocol cannot detect problems during the audit;
- Combined medications were not recorded;
- The storage conditions of the clinical trial samples were inconsistent with the actual conditions;
- The applicable model of the comparative reagent did not match the experiment instrument.
Clinical trial data management
- The records in the original medical records were incomplete;
- The image evaluation records related to clinical trials were incomplete;
- The original records of the clinical trial cases did not record the reasons and time of the modification;
- There was no screening record for the sample;
- The project training record was incomplete or missing;
- The audit record was incomplete or missing;
- The case report form records did not meet the requirements;
- The data in the case report form was inconsistent with the original record;
- The clinical trial test data was not confirmed by the operator or the reviewer;
- The adverse events, serious adverse events, equipment defects were missing;
- There was no basis or plausible reason for data deletion;
- The expiration date of the test product was not recorded;
- The use record of equipment for clinical trials was missing.
Management of test products
- The type test report was not submitted before conducting clinical trial;
- The distribution and recovery records of the tested products and control products were not provided;
- The tracking number was not provided by delivery record;
- The test sample reagent had expired when delivering to the clinical trial sites.
Management of samples
- Instruction of reused samples was not provided;
- The records of sample collection, storage, distribution, use, sample retention, and destruction were not incomplete or missing;
- The sample types are inconsistent, and samples are not traceable.
Application materials
- The protocol kept by the ethics committee was inconsistent with the number of cases and the distribution of cases in the registration application materials;
- The clinical trial protocol and report in the submitted registration application were inconsistent with the clinical trial protocol and report signature kept by the clinical trial institute;
- The clinical trial products were inconsistent with that in the clinical trial protocol and test report of the registration application materials;
- The clinical data were inconsistent with the statistical analysis data provided on-site;
- The statistical analysis report form did not provide plausible reasons for the case deletion;
- The number of follow-up cases kept by clinical sites was inconsistent with number of follow-up cases in the Clinical Case Report (CCR).
For our slides on Clinical Trials in China that we shared on RAPS annual meeting, please email info@ChinaMedDevice.com.
For our post on Suggestions for Clinical Auditing in China, please click HERE.
For our webinar on Key Ingredients for Effective China SDA (CFDA) and Clinical Trial, please click HERE.
Keep yourself updated with NMPA (CFDA) News Roundup, click HERE to opt-in. For market access newsletter, click HERE. For CRO services in China, click HERE.
About ChinaMed Device, LLC
ChinaMed Device, LLC (www.ChinaMedDevice.com) provides regulatory and commercialization turnkey solutions for western medical device, IVD, CDx and combination products to enter China. As a certified NMPA (CFDA) legal agent with offices in Boston and Beijing, we can represent overseas manufacturers for the complete product life cycle without their need to set up local entity in China. Our NMPA (CFDA) regulatory services include: RA, regulatory strategy, regulatory submissions, clinical evaluation reports (CER), clinical trials, QA, GMP and post-market compliance. Our commercialization services include: market assessment research, reimbursement, partnership strategies, and distribution qualification.